医用化验和基础设备(医疗器械检查内容)
- 电商
- 2022-06-27 22:44:43
医疗器械总共分为三类,第一类、第二类和第三类有什么区别?
根据*新《医疗器械监督管理条例》(国务院令第650号)和《医疗器械经营质量管理规范》首先要明白医疗器械分为三类:第一类、第二类、第三类。目前经营二类产品不需要办理《医疗器械经营许可证》,只需要办理二类医疗器械注册证,经营医疗器械的只有三类产品。然后,医疗器械经营企业申请第二类医疗器械经营备案和第三类医疗器械经营许可证。第一类是低风险的医疗器械,日常管理可以保证其安全性和有效性。如手术器械(刀、剪、钳、镊子、钩)、刮痧板、医用x光片、手术衣、手术帽、检查手套、纱布绷带、引流袋等。第二类是中等风险的医疗器械,需要严格控制和管理,以确保其安全性和有效性。如:手术器械、6815注射穿刺器械、6820普通诊断器械、6821医用电子仪器、6822医用光学仪器及内镜设备、6823医用超声仪器及相关设备、6824医用激光仪器、6825医用高频仪器、6826理疗康复设备、6827中医仪器、6828医用磁共振设备、6830医用6831医用X射线附属设备及部件、6833医用核素设备、6833 6840临床实验室分析仪器(体外诊断试剂限于早孕试纸、排卵试纸、尿糖试纸、血糖试纸)、6841医学实验室及基础设备、6845体外循环及血液处理设备、6846植入材料及人工器官、6854手术室、急诊室、诊室设备及器具、6855口腔科设备及器具、6856病房护理设备及器具、6857消毒灭菌设备及器具、6858医疗冷疗、 低温及制冷设备及器具、6863牙科材料、6864医用卫生材料及敷料、6865医用缝合材料及粘合剂、6866医用高封闭分子材料及制品、6870软件二类医疗器械备案经营范围。 第三类是高风险的医疗器械,需要采取特殊措施进行严格控制和管理,以保证其安全性和有效性,如:6804眼科手术器械;807胸心血管外科手术器械;810矫形外科(骨科)手术器械;615注射穿刺仪;821医用电子仪器及设备;822医用光学仪器、器械和内窥镜设备;822-1医用光学器械、仪器和内窥镜设备;823医用超声仪器及相关设备;824医用激光设备;825医用高频仪器设备;6826理疗设备6828医用磁共振设备;830台医用x光设备;831医用X射线附属设备及部件;832医用高能射击设备;833医用核素设备;840临床实验室分析仪器(不包括体外诊断试剂);845体外循环和血液处理设备;846植入材料和人造器官;手术室、急诊室和诊所的设备和器具654件;858医用冷疗、低温和制冷设备及器具;口腔医学663材料;865医用缝合材料及粘合剂;666医用高分子材料及制品;870软件;介入设备677台;826物理治疗和康复设备。
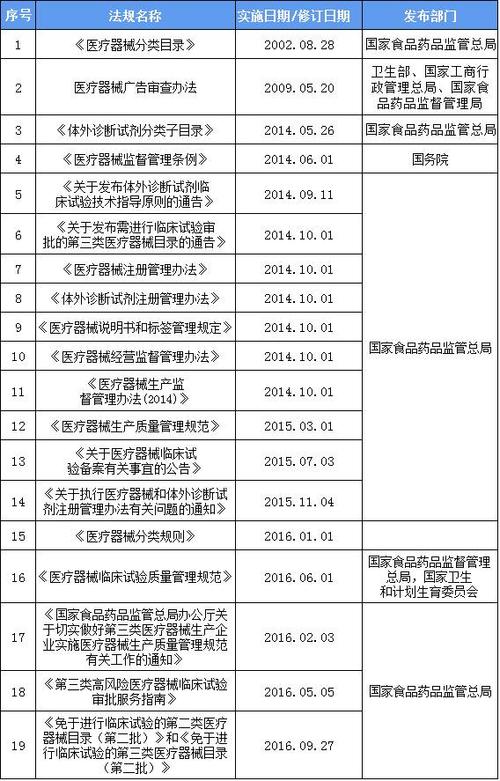
一类医疗器械的医疗器械分类目录
医疗器械分类目录:1。基础外科手术器械、显微外科手术器械、神经外科手术器械、眼科手术器械、耳鼻喉科手术器械、口腔科手术器械、腹部手术器械、泌尿肛肠手术器械、骨科(整形)手术器械、妇产科手术器械、计划生育手术器械、注射穿刺手术器械和烧伤(整形)手术器械、8。普通诊断设备、医用电子设备9、医用光学设备、仪器及内镜设备、医用超声设备及相关设备10、医用激光设备、医用高频设备11、理疗康复设备、中医设备12、医用磁共振设备、医用X射线设备13、医用X射线附件及部件、医用高能射线设备14、医用核素设备、医用辐射防护用品及装置15、临床分析仪器、医学实验室及基础设备和器具16、 体外循环和血液处理设备、植入材料和人造器官17、手术室、急诊室和诊所的设备和用具、口腔科的设备和用具18、病房护理设备和用具中的消毒灭菌设备和用具19、医用冷疗、低温和冷藏设备和用具、口腔科材料20、医用卫生材料和敷料、医用缝合材料和粘合剂21、 介入器械用医用高分子材料及制品扩展资料: (一)医疗器械备案是指食品药品监督管理部门将医疗器械备案人(以下简称备案人)提交的一类医疗器械备案资料进行备案备查。 (2)备案的医疗器械为《第一类医疗器械目录》及相应体外诊断试剂分类子目录中的第一类严复医疗器械。中国境内第一类医疗器械的备案,备案人应当向设区的市级食品药品监督管理部门提交备案材料。进口第一类医疗器械应当进行备案,备案人应当向中国食品药品监督管理局提交备案材料。香港、澳门和台湾地区医疗器械的备案参照进口医疗器械办理。(三)备案的进口医疗器械,应当在备案人注册的国家(地区)或者生产地址所在地取得医疗器械上市许可。产品在注册地或备案人生产地址所在国家(地区)未作为医疗器械管理的,备案人应提供相关证明文件,包括备案人注册地或生产地址所在国家(地区)允许该产品合法上市销售的证明文件。(四)境外备案人在进口医疗器械备案时,应当通过其在中国境内设立的代表机构或者委托中国境内的企业法人作为代理人办理。(五)备案人应当准备好拟备案医疗器械的产品技术要求。技术要求主要包括医疗器械成品的性能指标和检验方法。(6)
)办理医疗器械备案,备案人应当按照相关要求(见附件1)提交备案资料,并对备案资料的真实性、完整性、合规性负责。(七)备案资料符合要求的,食品药品监督管理部门应当当场予以备案。备案资料不齐全或者不符合规定形式的,应当一次告知需要补正的全部内容。对不予备案的,应当告知备案人并说明理由。(八)对予以备案的医疗器械,食品药品监督管理部门应当按照相关要求的格式制作备案凭证(见附件2),并将备案信息表(见附件3)中登载的信息在其网站上予以公布。食品药品监督管理部门按照第一类医疗器械备案操作规范(见附件4)开展备案工作。备案人应当将备案号标注在医疗器械说明书和标签中。(九)已备案的医疗器械,备案信息表中登载内容及备案的产品技术要求发生变化,备案人应当提交变化情况的说明及相关证明文件,向原备案部门提出变更备案信息。食品药品监督管理部门对备案资料符合形式要求的,应在变更情况栏中载明变化情况,将备案资料存档。参考资料来源:百度百科_一类医疗器械
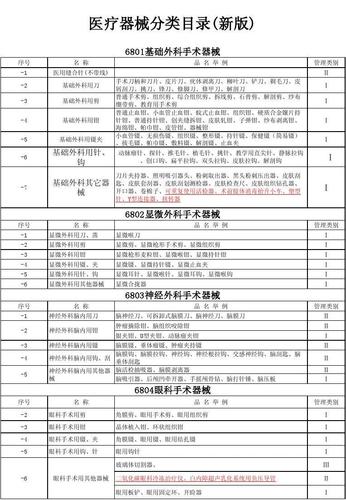
医疗器械分类包括什么?
医疗器械分类包括:第一类为通过常规管理足以保证其安全性、医学|教育|网搜集整理有效性的医疗器械。如大部分手术器械、听诊器、医用x线胶片、医用X线防护装置、全自动电泳仪、医用离心机、切片机、牙科椅、煮沸消毒器、纱布绷带、弹力绷带、橡皮膏、创可贴、拔罐器、手术衣、手术帽、口罩、集尿袋等。第二类为对其安全性、有效性应当加以控制的医疗器械。如体温计、血压计、助听器、制氧悉岩档机、避孕套、针灸针、心电诊断仪器、无创监护仪器、光学内窥镜、便携式超声睁乱诊断仪、全自动生化分析仪、恒枣枣温培养箱、牙科综合治疗仪、医用脱脂棉、医用脱脂纱布等。第三类用于植入人体或支持维持生命,对人体具有潜在危险,对其安全性、有效性必须严格控制的医疗器械。如植入式心脏起搏器、体外震波碎石机、病人有创监护系统、人工晶体、有创内窥镜、超声手术刀、彩色超声成像设备、激光手术设备、高频电刀、微波治疗仪、医用核磁共振成像设备、x线治疗设备、200mA以上x线机、医用高能设备、人工心肺机、内固定器材、人工心脏瓣膜、人工肾、呼吸麻醉设备、一次性使用无菌注射器、一次性使用输液器、输血器、CT设备等。
- 人参与,0条评论
发表评论